1901年,德国精神病学家Alois Alzheimer对一位患者进行尸检,银染后观察到神经元内和神经元之间存在两种类型的沉积物1。他将其描述为“大脑皮层的一种特殊疾病”——这便是阿尔茨海默病(Alzheimer’s Disease, AD)。AD是一种以淀粉样斑块沉积和神经纤维缠结为特征的进行性神经退行性疾病,是最常见的痴呆形式。9月21日是世界阿尔茨海默病日。在这个特殊的日子,让我们一起走近阿尔茨海默病,了解如何选择AD动物模型吧。
AD发病机制及风险基因
任何疾病的发生都离不开遗传因素和环境因素的共同作用,阿尔茨海默病也不例外。双胞胎研究表明,患阿尔茨海默病的风险有60-80%取决于遗传因素2。
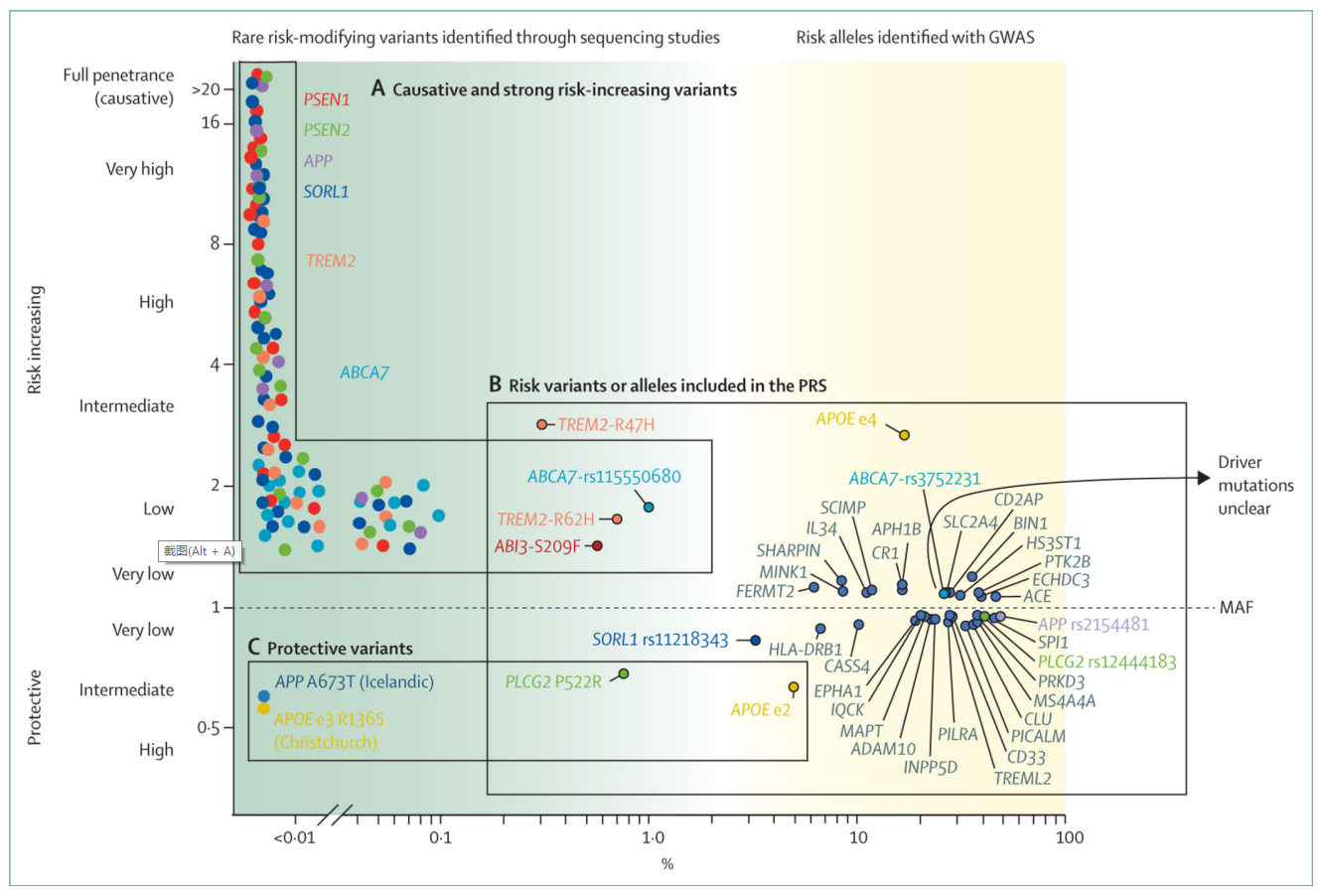
阿尔茨海默病基因图谱2
横轴是群体中相应基因突变出现的频率,纵轴则代表患有阿尔茨海默病的风险。OR=1表示该突变的携带者与非携带者患阿尔茨海默病的几率相同,OR>1表示突变可增加患阿尔茨海默病风险,OR<1则表示突变具有保护作用。
虽然AD的发病机制目前尚不清楚,但整体而言,AD涉及多种致病原因和多种中枢神经系统细胞类型3。β淀粉样蛋白(β-amyloid, Aβ)由神经元产生,随后被释放至细胞外基质,可被小胶质细胞和星形胶质细胞清除并降解。Aβ的合成增加,或者Aβ的降解受损,都有可能导致Aβ的累积。Tau蛋白主要在神经元中表达,并且存在着多种翻译后修饰(post-translational modifications, PTMs)。异常PTM、液相分离(liquid-liquid phase separation, LLPS)和致病性Tau可通过不同的机制引起Tau聚集和积累。各种形式的Aβ聚集物和Tau蛋白可以引起神经元功能障碍和胶质细胞激活,随后导致神经炎症。
Aβ和AD
Aβ是AD相关淀粉样斑块的主要成分,由I型跨膜淀粉样前体蛋白(amyloid precursor protein, APP)经蛋白酶裂解产生。根据产生的裂解产物不同,APP加工可分为非淀粉样蛋白加工途径和淀粉样蛋白加工途径4。
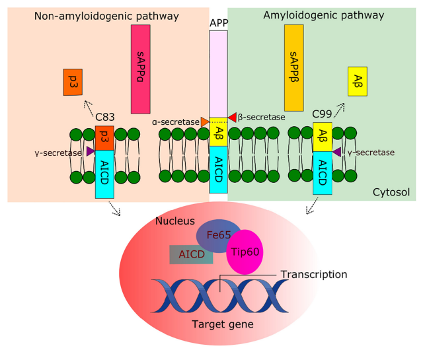
APP剪切过程4
根据是否产生Aβ,APP的剪切过程可分为两类:β-和γ-分泌酶介导淀粉样蛋白加工途径,α-和γ-分泌酶介导非淀粉样蛋白加工途径。在非淀粉样蛋白加工途径中,APP在α-分泌酶催化下产生sAPPα和αCTF,随后被γ分泌酶裂解产生p3和AICD。由于α分泌酶的剪切位点位于Aβ序列之内,因此该剪切途径不会产生Aβ。在淀粉样蛋白形成过程中,β-分泌酶介导APP剪切产生sAβ和β-CTF,随后β-CTF被γ分泌酶裂解产生Aβ和AICD。
淀粉样蛋白加工途径包括β-分泌酶和γ-分泌酶对APP蛋白的依次水解。Aβ42和Aβ40是大脑中两种主要的Aβ。由于Aβ42具有聚集倾向,并被证明具有神经毒性,因此Aβ42被认为是启动淀粉样斑块形成和AD发病机制的关键成分。
人类Tau蛋白由17号染色体上的MAPT基因编码,具有多种同源异构体。Tau蛋白存在多种PTM,包括磷酸化、泛素化、乙酰化、甲基化等,这些PTM可影响Tau蛋白的聚集。以磷酸化为例,在AD发展的早期阶段,患者脑脊液中就可检测到磷酸化Tau的水平升高5。生理状态下,Tau蛋白可以作为微管结合成分,促进微管的聚合和稳定性。磷酸化可影响Tau与微管结合:Tau蛋白多个位点过度磷酸化可导致Tau与微管分离,并增强Tau聚集6。
APOE和AD
ApoE是中枢神经系统中主要的脂质和胆固醇载体。人类有三种主要的APOE等位基因,分别为ε2 (APOE2)、ε3 (APOE3)和ε4 (APOE4)。APOE4可以基因剂量依赖的方式增加AD的风险,APOE4纯合人群患有AD的风险升高15倍;而APOE2则可降低AD风险,并与寿命增加有关7。除不同的等位基因外,APOE还存在着多种多样的突变类型,一同影响患有AD的风险。虽然APOE与AD风险有关已是学界共识,但其并非导致AD的风险基因;APOE在AD中发挥的作用也有待后续研究8,9。
TREM2和AD
TREM2是一种属于免疫球蛋白超家族的单向跨膜免疫反应受体,在中枢神经系统中主要由小胶质细胞表达。TREM2可与DAP10或DAP12结合并形成异源二聚体,介导下游信号传导。虽然TREM2的功能尚不完全清楚,但已经发现在中枢神经系统中,TREM2可调节炎症信号传导和小胶质细胞代谢,并能促进小胶质细胞吞噬、活化、存活和增殖10。最常见的TREM21 R47H突变可使AD风险增加2-3倍,而其它TREM2突变也可通过影响TREM2表达、表面转运、配体结合或信号转导等方面增加AD风险11。近日研究表明,应用TREM2拮抗性抗体后,可加重5×FAD小鼠中小胶质细胞激活、tau病理沉积以及神经营养不良情况,但不影响Aβ斑块表型12。
根据AD的发病原理,研究者们构建了多种多样的动物模型。根据ALZFORUM | NETWORKING FOR A CURE数据库收录,目前可用于进行AD相关研究的小鼠模型就有近200种。在这些动物模型中,最常见的就是基因工程相关品系。另外,使用自然衰老或加速衰老小鼠、注射腺相关病毒(adeno-associated virus, AAV)、致病蛋白或预制纤维(preformed fibrils, PFFs)等都是常用的造模方式。但是,目前为止,没有一种小鼠能够完全反映人类AD的各个方面。因此,在正式开展AD相关研究时,您需要慎重选择合适的动物模型,让您的研究事半功倍。那么我们一起来看看,在选择基因工程AD动物模型时,需要考虑哪些因素吧。
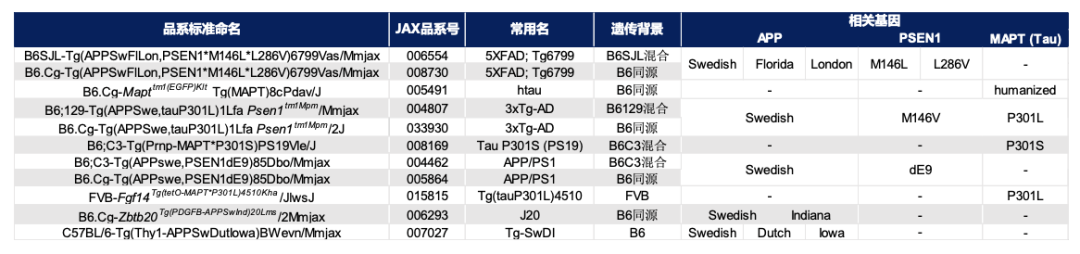
APP、PSEN1等基因已被明确为AD的致病基因,并且基于这些致病基因构建的各种动物模型,都能够在不同程度上复现AD的表型。但是,不同基因突变导致AD表型,其机制很可能各不相同。因此,在研究开始前,您需要考虑在研究中是否会涉及到特定基因。我们总结了JAX最受欢迎的11种AD动物模型相关的基因及突变类型,您可以参照表格进行选择。
以APP基因为例,在人群中这一基因可能存在不同的突变类型,常见的有瑞典突变(APPswe: K670N/M671L)、伦敦突变(APPlon: V717I)和印第安纳突变(APPind: V717F)等。不同的突变导致Aβ聚集的原因又各不相同13:假设突变发生在β剪切酶切割位点(例如瑞典突变),会造成β剪切的增加;假设突变发生在γ剪切酶切割位点(例如伦敦突变),则导致Aβ产生的增加。若您想以抑制β-分泌酶、减少β剪切为策略研发治疗AD的新药物,那么携带有APPswe突变的相关品系可能更适合您的研究。
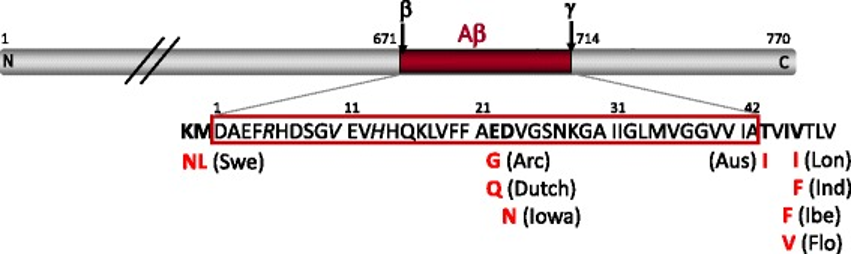
APP常见突变类型14
图中展示了Aβ的氨基酸序列,箭头标注β-和γ分泌酶的切割位点。图中粗体表示常见的FAD突变位点及其名称。
此外,多项大规模的全基因组关联分析(genome-wide association study, GWAS)及Meta分析均发现,APOE ε4等位基因仍然是散发性AD最强的遗传危险因素,APOE ε2等位基因则是最强的遗传保护因素14。若您需要研究APOE与AD的关系,JAX也提供以下活体品系供您选择。
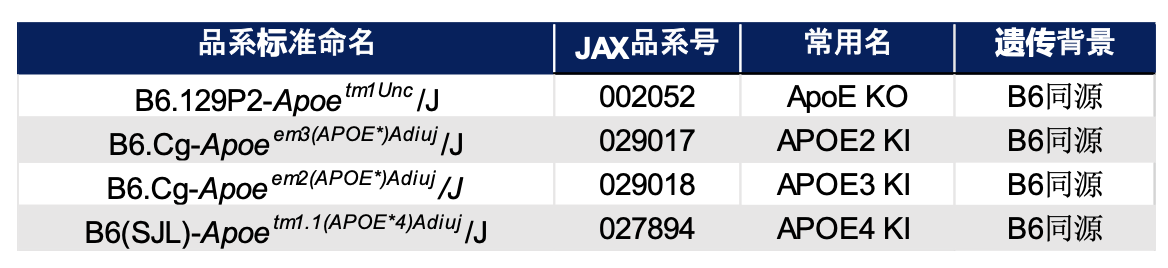
JAX 可提供APOE活体品系
疾病表型
正如先开始所提到,AD最常见的疾病表型可以分为包括病理改变和行为学异常等在内的多个方面,病理改变又可以划分为淀粉样斑块、神经纤维缠结、胶质细胞激活和神经元丢失等。在研究开始之前,您需要考虑您想观察AD动物模型的哪些方面,检测指标又是什么?这些需要您进行大量的文献调研,但您也可以考虑使用数据库帮助节省您的时间。ALZFORUM | NETWORKING FOR A CURE数据库不仅收录了AD动物模型,也同时收录了这些模型的部分表型。基于ALZFORUM数据库中的记录,我们梳理了JAX最受欢迎的10种AD动物模型的表型。
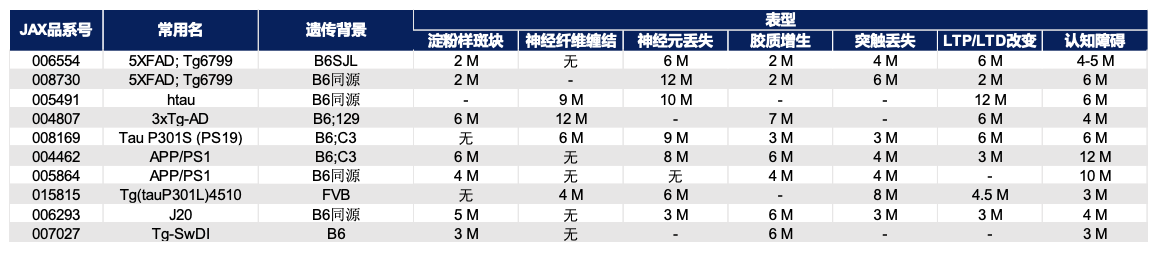
JAX 常用AD动物模型表型总结16;“-”表示暂无数据
假设您主要想研究的是某一分子或者某一药物是否能改善AD动物模型中异常积聚的tau蛋白,那在选择模型时,需要考虑感兴趣的动物模型是否能够出现tau蛋白水平升高和神经纤维缠结等表型。需要提醒您的是,由于环境等因素很可能会影响小鼠出现表型的时间以及表型的严重程度,检测手段及观测指标也可能部分影响结论的得出(特别是行为学结果),其他研究者得到的结果很可能与在您设施中观察到的不同。因此,在开展正式实验前,记得选取部分动物进行预实验哦。
遗传背景
具有相同突变但遗传背景不同的小鼠,表型可能有所差异。以APP/PS1小鼠为例,目前JAX可以提供两个品系:
004462 - APP/PS1,B6;C3混合遗传背景;
005864 - APP/PS1,B6同源遗传背景。
B6同源遗传背景的005864品系小鼠是使用004462品系小鼠与C57BL/6J回交得到的,这两个品系转入的基因相同,遗传背景不同。然而,这两种品系小鼠在发生癫痫 的概率上存在着巨大的差异:B6同源遗传背景的005864品系小鼠在4.5月龄之前癫痫发生率为55%17;但是B6;C3混合遗传背景的004462品系小鼠则没有癫痫的表型。另外,根据ALZFORUM数据库中的记录,B6同源遗传背景的005864品系小鼠出现淀粉样斑块的时间早于B6;C3混合遗传背景的004462品系小鼠;并且根据数据库中的记录,B6同源遗传背景的005864品系小鼠不会出现神经元丢失表型。因此,若想使用APP/PS1小鼠研究淀粉样斑块表型,选择B6同源遗传背景的品系可能更有利于加速研究进程;若想研究神经元丢失表型,则B6;C3混合遗传背景的004462品系小鼠更符合实验需求。
研究进程
假如您在考虑上述问题后,仍无法确定您需要的品系,您还可以从影响您研究进程的其它因素进行考量。在进行配繁时,您需要制定合适的配繁方案,以尽快产生足够数目的实验小鼠(关于如何制定合适的繁育策略,您可参看小鼠基础知识十一:制定合适的繁育策略,让你的研究更高效。以5×FAD小鼠为例,目前JAX可以提供两个品系:
006554 - 5XFAD ,B6SJL混合遗传背景;
008730 - 5xFAD , 5x-FAD, Tg6799,B6同源遗传背景;
若您考虑使用B6SJL混合遗传背景的品系(#006554),为了维持遗传背景的稳定,您需要使用该品系半合子小鼠与B6SJLF1/J (#100012)小鼠 进行配繁。B6SJLF1/J (100012)品系不能自我维持,只能通过雌性C57BL/6J (#000664)×雄性SJL (#000686)来获得,因此繁育该5×FAD小鼠需要维持三个种群,相对较为复杂。
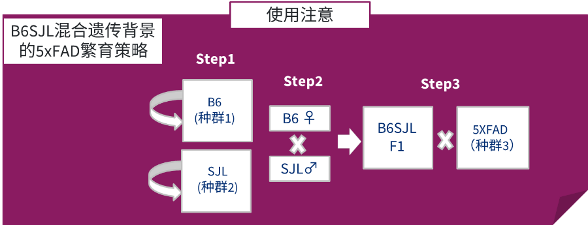
B6SJL混合遗传背景的5×FAD小鼠繁育策略
另外, AD动物模型相对较长的造模时间也给研究者们带来了挑战。假设您想研究的AD主要表型为神经元丢失,参考上面的表型总结,选择更早出现神经元丢失表型 的B6.Cg-Zbtb20Tg(PDGFB-APPSwInd)20Lms/2Mmjax (#006293, J20)品系小鼠可能比B6.Cg-Tg(APPSwFlLon,PSEN1*M146L*L286V)6799Vas/Mmjax (#008730, 5×FAD)品系小鼠更有利于高效开展研究。
动物来源
当确定您所需要使用的品系后,您就可以开始着手于动物引进。动物的来源有很多,您既可以联系其他实验室有无您感兴趣的品系,寻求捐赠,也可以从可靠的供应商处购买获得(例如JAX、MMRRC和EMMA)。虽然从其他实验室获得动物较为快速且便宜,但其遗传质量较难得到保障。例如在JAX,会对多个品系进行单核苷酸多态性(SNP)分析,以明确品系的遗传背景。另外,J20小鼠品系被报导存在转基因拷贝数丢失的情况后,JAX进行了重新引种,并利用qPCR监测转基因拷贝数目,以确保您能获得高质量的动物模型。
通过这篇文章,相信大家对AD的发病机理以及如何选择AD转基因动物模型已有初步的了解。除经典AD动物模型外,研究者们也致力于开发更能模拟晚发型AD的动物模型,您可点击Model AD | (model-ad.org)查看相关信息。杰克森实验室先前也推出了视频版本的阿尔茨海默症小鼠模型介绍,您也可以点击视频号|阿尔兹海默症小鼠模型,助你找回“消失”的时间 查看。
若您对AD疾病研究及药物研发感兴趣,可点击品系查询寻找适合的AD动物模型;您也可参照JAX®小鼠模型搜索指南 进行检索。我们强烈建议您在选择小鼠品系前仔细阅读小鼠的品系详情,并查看小鼠品系相关的参考文献,进行全面的文献调研,确保您选择的小鼠模型能够满足您的实验需求。您也可以联系杰克森实验室技术支持(micetech@jax.org.cn, 400-001-2626)获取更多相关信息,我们将竭尽所能为您提供帮助。
参考文献
1. Morris RG, Salmon DP. “The centennial of Alzheimer's disease and the publication of "Uber eine eigenartige Erkankung der Hirnrinde" by Alöis Alzheimer”. Cortex. 2007 Oct;43(7):821-5. doi: 10.1016/s0010-9452(08)70681-6. PMID: 17941340.
2. Scheltens P et al. “Alzheimer's disease”. Lancet. 2021 Apr 24;397(10284):1577-1590. doi: 10.1016/S0140-6736(20)32205-4. Epub 2021 Mar 2. PMID: 33667416; PMCID: PMC8354300.
3. Guo T et al. “Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer's disease”. Molecular Neurodegeneration. 2020 Jul 16;15(1):40. doi: 10.1186/s13024-020-00391-7. PMID: 32677986; PMCID: PMC7364557.
4. Cho Y et al. “Physiology and pharmacology of amyloid precursor protein”. Pharmacology & Therapeutics. 2022 Jul;235:108122. doi: 10.1016/j.pharmthera.2022.108122. Epub 2022 Feb 1. Erratum in: Pharmacol Ther. 2022 Oct;238:108281. PMID: 35114285.
5. Bai B et al. “Proteomic landscape of Alzheimer's Disease: novel insights into pathogenesis and biomarker discovery”. Molecular Neurodegeneration. 2021 Aug 12;16(1):55. doi: 10.1186/s13024-021-00474-z. Erratum in: Mol Neurodegener. 2021 Oct 20;16(1):72. PMID: 34384464; PMCID: PMC8359598.
6. Xia Y et al. “"Don't Phos Over Tau": recent developments in clinical biomarkers and therapies targeting tau phosphorylation in Alzheimer's disease and other tauopathies”. Molecular Neurodegeneration. 2021 Jun 5;16(1):37. doi: 10.1186/s13024-021-00460-5. PMID: 34090488; PMCID: PMC8180161.
7. Raulin AC et al. “ApoE in Alzheimer's disease: pathophysiology and therapeutic strategies”. Molecular Neurodegeneration. 2022 Nov 8;17(1):72. doi: 10.1186/s13024-022-00574-4. PMID: 36348357; PMCID: PMC9644639.
8. Martens YA et al. “ApoE Cascade Hypothesis in the pathogenesis of Alzheimer's disease and related dementias”. Neuron. 2022 Apr 20;110(8):1304-1317. doi: 10.1016/j.neuron.2022.03.004. Epub 2022 Mar 16. PMID: 35298921; PMCID: PMC9035117.
9. Fernández-Calle R et al. “APOE in the bullseye of neurodegenerative diseases: impact of the APOE genotype in Alzheimer's disease pathology and brain diseases”. Molecular Neurodegeneration. 2022 Sep 24;17(1):62. doi: 10.1186/s13024-022-00566-4. PMID: 36153580; PMCID: PMC9509584.
10. Li RY et al. “TREM2 in the pathogenesis of AD: a lipid metabolism regulator and potential metabolic therapeutic target”. Molecular Neurodegeneration. 2022 Jun 3;17(1):40. doi: 10.1186/s13024-022-00542-y. PMID: 35658903; PMCID: PMC9166437.
11. Hou J et al. “TREM2 dependent and independent functions of microglia in Alzheimer's disease”. Molecular Neurodegeneration. 2022 Dec 23;17(1):84. doi: 10.1186/s13024-022-00588-y. PMID: 36564824; PMCID: PMC9783481.
12. Jain N et al. “Chronic TREM2 activation exacerbates Aβ-associated tau seeding and spreading”. Journal of Experimental Medicine. 2023 Jan 2;220(1):e20220654. doi: 10.1084/jem.20220654. Epub 2022 Oct 11. PMID: 36219197; PMCID: PMC9559604.
13. Karran E et al. “The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics”. Nature Reviews Drug Discovery. 2011 Aug 19;10(9):698-712. doi: 10.1038/nrd3505. PMID: 21852788.
14. Serrano-Pozo A et al. “APOE and Alzheimer's disease: advances in genetics, pathophysiology, and therapeutic approaches”. The Lancet Neurology. 2021 Jan;20(1):68-80. doi: 10.1016/S1474-4422(20)30412-9. Erratum in: Lancet Neurol. 2021 Feb;20(2):e2. PMID: 33340485; PMCID: PMC8096522.
15. Jankowsky JL, Zheng H. “Practical considerations for choosing a mouse model of Alzheimer's disease”. Molecular Neurodegeneration. 2017 Dec 22;12(1):89. doi: 10.1186/s13024-017-0231-7. PMID: 29273078; PMCID: PMC5741956.
16. Alzheimer's Disease Research Models | ALZFORUM
17. Minkeviciene R et al. “Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy”. Journal of Neuroscience. 2009 Mar 18;29(11):3453-62. doi: 10.1523/JNEUROSCI.5215-08.2009. PMID: 19295151; PMCID: PMC6665248.
