2014年,国际肌营养不良家长联盟(United Parent Muscular Dystrophy)组织首次将每年的9月7日定为DMD世界知晓日(World Duchenne Awareness Day, WDAD),旨在让更多人知道杜氏肌营养不良,让更多人了解患者的真实生活。那么,你知道什么是DMD吗?
杜氏肌营养不良(Duchenne muscular dystrophy, DMD)是一种严重的进行性肌肉萎缩疾病,每5000-6000名新生男婴中就有一人患病。该病由位于X染色体上的编码抗肌萎缩蛋白(Dystrophin)的DMD基因突变导致,呈X染色体隐性遗传1。在DMD患者中,有60-70%为缺失突变,5-15%为重复突变,而其余20%为点突变、小的删除或插入1。DMD基因的缺失和重复突变集中于45-55和3-9号外显子,分别有47%和7%的DMD患者携带这两处的突变1。DMD的发病具有明显的性别差异:女性具有两条X染色体,因此鲜有女性患DMD。生理状态下,Dystrophin可以连接纤维型肌动蛋白(F-actin)和细胞外基质,同时帮助维持肌细胞的稳定性,使得肌细胞在收缩过程中免受破坏。该基因突变后,由于无法产生Dystrophin的肌肉异构体,患者在2-3岁时就表现出经常跌倒、蹒跚步态等早期症状。随着疾病的不断进展,患者通常于40岁前因心脏功能障碍或呼吸衰竭死亡。
由于DMD起病隐匿,当父母发现儿童出现运动异常时,疾病已进展了数月甚至数年。但令人振奋的是,随着对于DMD的认识不断深入,新药研发为DMD的对症治疗甚至治愈提供了希望2。下面让我们一起看看目前已获批可用于治疗DMD的药物,它们采用的治疗策略以及在药物研发过程中使用的动物模型吧。
基因疗法
今年6月22日,Serepta Therapeutics公司宣布3,其与Roche联合开发的基因疗法SRP-9001(Elevidys)获得美国食品药品监督管理局(Food and Drug Administration, FDA)加速批准上市,成为首个用于治疗4-5岁DMD患者的基因疗法——这是自2017年来第13个获得FDA批准的基因疗法药物,也是首个针对儿童遗传疾病的基因疗法。
获批的SRP-9001是由Sarepta公司所生产的一种基因治疗药物,罗氏(Roche)公司拥有其在美国以外地区的独家权力。由于DMD基因的长度大于2Mb,超过基因治疗使用的腺相关病毒(Adeno-associated virus, AAV)载体所能递送的范围,因此研究人员通过只恢复基因最重要部分的方式来解决这个问题。SRP-9001原理是通过单次静脉注射,将编码微型抗肌萎缩蛋白(Micro-dystrophin)的转基因递送至肌肉组织,以代替Dystrophin的主要功能4。
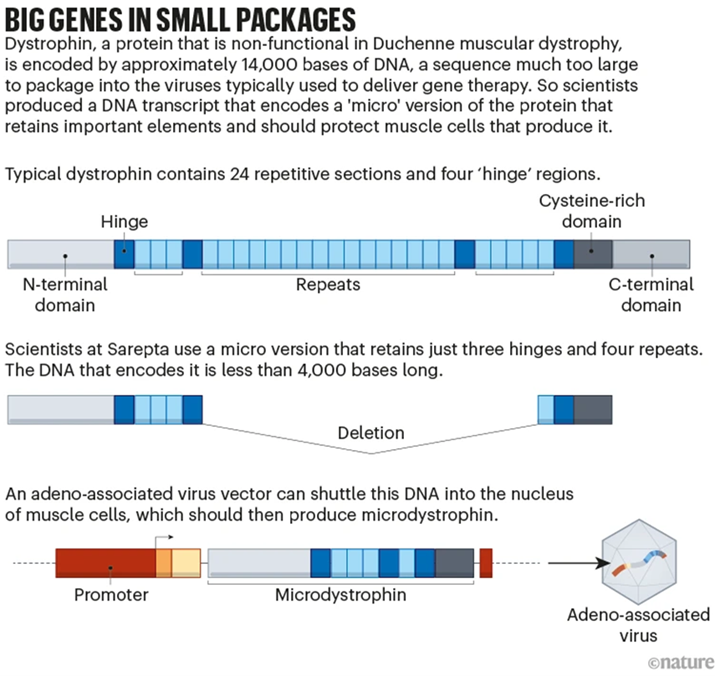
SRP-9001构建策略4
SRP-9001及其前体药物的研发始于2005年。在临床前研究中,研究者使用了C57BL/10ScSn-Dmdmdx/J小鼠(#001801,通常称为mdx小鼠)进行药效评估。mdx小鼠于1984年由Thomas Roderick博士引入杰克森实验室。由于Dmd基因第23号外显子内C到T的自发突变,这些小鼠并不表达Dystrophin,因此可用于研究DMD。mdx小鼠从3周左右开始出现肌肉坏死以及肌肉无力表型,相关的病理性生化改变包括血清肌酸激酶(Creatine kinase, CK)和丙酮酸激酶(Pyruvate kinase, PK)水平升高,以及巨噬细胞的积累——二者都是肌肉变性的早期标志。在使用SRP-9001后,实验者观察到β-肌聚糖在肌膜上的定位增加、循环CK显著减少及mdx小鼠胫骨前肌和膈肌的比力输出(Specific force output)增加的现象5,标志着DMD动物的肌萎缩情况得到改善,提示SRP-9001在动物上取得了较好的治疗效果。随后,该药物于2018年1月开始1期临床试验(NCT03375164)。使用SRP-9001后,患者循环CK显著减少且肌肉功能得到显著改善。并且在治疗后的四年中,受试者的其他运动功能指标也普遍得到了改善6,7。
“通读”(readthrough)是指核糖体忽视终止密码子继续进行翻译的现象。利用这一机制,PTC Therepeutics公司研发了Ataluren,可以使得在蛋白翻译过程中绕过DMD基因无义突变而产生的提前终止密码子8。
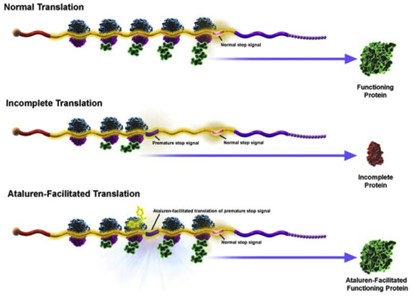
Ataluren作用机制9
在Ataluren的临床前研究中,研究者同样使用了mdx小鼠(#001801)。在表达无义Dystrophin等位基因的人及mdx小鼠原代肌细胞中,PTC124可促进Dystrophin蛋白的产生,并在2-8周内恢复mdx小鼠的横纹肌功能。目前,Ataluren正在6个月至2岁无义突变DMD患者中开展安全新及药代动力学研究(NCT04336826)。
外显子跳跃
反义寡聚核苷酸(Antisense oligonucleotide, ASO)介导的外显子跳跃被认为是目前最有前景的DMD治疗方法之一2。该治疗方法可利用针对DMD前体mRNA的ASO,跳过至少一个外显子,从而使得读码框恢复正常。在SRP-9001获批前,全球已有多款治疗DMD的“外显子跳跃”药物,包括Sarepta Therapeutics公司研发的Amondys 45(Casimersen)、Exondys 51(Eteplirsen)和Vyondys 53(Golodirsen),以及Nippon Shinyaku公司研发的Viltepso(Viltolarsen)。
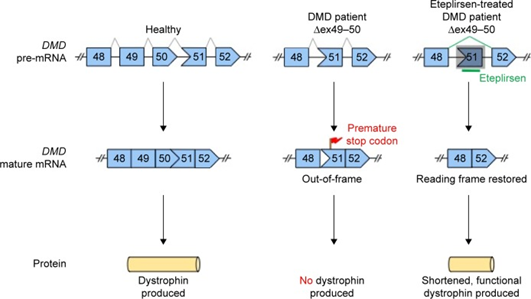
Eteplirsen治疗原理10
以首个获批用于DMD的药物——Eteplirsen(先前称作AVI-4658)为例,为了开发针对人源DMD基因的ASO药物,研究者使用了转入完整人源DMD基因的转基因小鼠STOCK Tg(DMD)72Thoen/J(#018900,通常称为hDMD小鼠)。该品系小鼠具有功能性的人DMD基因,可用于针对DMD序列特异性疗法的临床前试验。研究者使用hDMD小鼠模型及培养的人肌肉细胞进行实验,对寡聚核苷酸条件进行了优化11,在评估安全性后开展临床研究。
除SRP-9001外,许多以递送micro-dystrophin为治疗策略的DMD基因疗法也在研发中,例如Solid Biosciences公司的SGT-001(NCT03368742);Capricor公司的细胞疗法CAP-1002(NCT05126758)等也受到广泛关注2。无论采用何种治疗策略,我们希望DMD这一疾病早日被人类攻克。
杰克森实验室的使命是为人类疾病探究、寻找精准的基因解决方案,赋能全球生物医药研究,为改善人类健康这一共同诉求做出贡献。目前,杰克森实验室可为全球提供超过13,000个遗传背景明确的精准小鼠模型。除上述mdx小鼠(#001801)外,JAX也提供D2.B10-Dmdmdx/J(#013141)、B6Ros.Cg-Dmdmdx-4Cv/J (#002378)、B10ScSn.Cg-Utrntm1Ked Dmdmdx/J (#019014)等DMD小鼠模型,希望能为药物研发做出贡献。您可点击链接进入小鼠品系详情页,或者参看品系查询,寻找合适的动物模型。同时,杰克森实验室也提供肌肉萎缩症体内药效研究服务。您可以联系杰克森实验室技术支持(micetech@jax.org.cn, 400-001-2626)获取更多相关信息,我们将竭尽所能为您提供帮助。
- Duan D et al. “Duchenne muscular dystrophy”. Nature Reviews Disease Primers. 2021 Feb 18;7(1):13. doi: 10.1038/s41572-021-00248-3. PMID: 33602943.
- Sheikh O, Yokota T. “Developing DMD therapeutics: a review of the effectiveness of small molecules, stop-codon readthrough, dystrophin gene replacement, and exon-skipping therapies.” Expert Opinion on Investigational Drugs. 2021 Feb;30(2):167-176. doi: 10.1080/13543784.2021.1868434. Epub 2021 Jan 6. PMID: 33393390.
- Sarepta Therapeutics Announces FDA Approval of ELEVIDYS, the First Gene Therapy to Treat Duchenne Muscular Dystrophy | Sarepta Therapeutics, Inc.
- Reardon S. “'It's a vote for hope': first gene therapy for muscular dystrophy nears approval, but will it work?”. Nature. 2023 Jun 2. doi: 10.1038/d41586-023-01799-z. Epub ahead of print. PMID: 37268837.
- Cellular, Tissue, and Gene Therapies Advisory Committee May 12, 2023 Meeting Briefing Document- Sponsor (fda.gov)
- Mendell JR et al. “Assessment of Systemic Delivery of rAAVrh74.MHCK7.micro-dystrophin in Children With Duchenne Muscular Dystrophy: A Nonrandomized Controlled Trial”. JAMA Neurology. 2020 Sep 1;77(9):1122-1131. doi: 10.1001/jamaneurol.2020.1484. PMID: 32539076; PMCID: PMC7296461.
- Potter RA et al. “Dose-Escalation Study of Systemically Delivered rAAVrh74.MHCK7.micro-dystrophin in the mdx Mouse Model of Duchenne Muscular Dystrophy”. Human Gene Therapy. 2021 Apr;32(7-8):375-389. doi: 10.1089/hum.2019.255. Epub 2021 Feb 18. PMID: 33397205; PMCID: PMC8063270.
- Welch EM et al. “PTC124 targets genetic disorders caused by nonsense mutations”. Nature. 2007 May 3;447(7140):87-91. doi: 10.1038/nature05756. Epub 2007 Apr 22. PMID: 17450125.
- Haas M et al. “European Medicines Agency review of ataluren for the treatment of ambulant patients aged 5 years and older with Duchenne muscular dystrophy resulting from a nonsense mutation in the dystrophin gene”. Neuromuscular Disorders. 2015 Jan;25(1):5-13. doi: 10.1016/j.nmd.2014.11.011. Epub 2014 Nov 24. PMID: 25497400.
- Lim KR, Maruyama R, Yokota T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Design Development and Therapy. 2017 Feb 28;11:533-545. doi: 10.2147/DDDT.S97635. PMID: 28280301; PMCID: PMC5338848.
- Arechavala-Gomeza V et al. “Comparative analysis of antisense oligonucleotide sequences for targeted skipping of exon 51 during dystrophin pre-mRNA splicing in human muscle”. Human Gene Therapy. 2007 Sep;18(9):798-810. doi: 10.1089/hum.2006.061. PMID: 17767400.
